Input formatting and examples¶
The input can be any text file, but sticking with .txt or .inp is recommended.
Any blank line will be ignored.
Comments (content of lines after
#) will be ignored.Keywords, if present, need to be on first non-blank line.
Then, molecule files are specified. FIRECODE works with .xyz files. A molecule line is made up of these elements, in this order:
Operators (i.e.
csearch>,opt>, etc.) separated by spacesThe molecule file name (required)
Indices (numbers) and pairings (letters) for the molecule (i.e.
2A 4B 5c). Letters are only used for legacy embeddings.Properties of the molecule (i.e.
charge=1,property=value)
Note
Molecule indices are zero-based! (counted starting from zero!)
By default, molecular charge is read from the filenames by counting the number of + and - characters.
However, this can be overridden by explicitly setting charge=n in the molecule line.
Uppercase letters specify fixed constraints (always enforced) while lowercase letters specify temporary
constraints. Temporary constraints are relaxed at the end of the refine> operator.
Constraints¶
A molecule line can be followed by one or more constraints: these lines should start with one or more
spaces. The line constraint syntax is {type} {i1} {i2} [{i3}] [{i4}] [{target} | "auto" | "ts"].
There are three types of constraints implemented: B (bonds), A (planar angles), and D (dihedrals).
These have to be followed by two, three or four indices, respectively. Then, an optional last parameter
specifies the target value. If a number is provided, this will be taken as the target distance in Å or angle
in degrees. Omitting this last input of specifying auto will use the current value read from the first
conformer. For bond constraints, specifying ts will set the target distance to 1.35 times the sum of
the elements’ covalent radii.
Constraint lines, like molecule lines, can also read properties. The only implemented for now is
“fixed” (default is fixed=true) mirroring the behavior of uppercase and lowercase letters in molecule line constraints.
Operators¶
The core elements of every run are the operators acting on a given molecule. See the operators page to see the full set of operators available. This should cater for most common workflow needs, from conformational search protocols to ensemble optimizations or double-ended TS-search methods like NEB or FSM.
Input examples¶
1. Conformational search and refinement¶
CALC=TBLITE LEVEL=GFN2-XTB SOLVENT=DMF T_C=-10
freq> opt> crest_search> ala_ala.xyz
# This is a comment line!
# First row sets the level of theory at the GFN2-XTB level via XTB.
# If XTB is not set as the default calculator, you can specify it
# here adding CALC=XTB in the keyword line.
# The operators on ala_ala.xyz are applied starting from the inside out:
# crest_search> - a metadynamics-based conformational search via CREST
# is carried out on ala_alat.xyz, generating ala_ala_crest_confs.xyz
# The default level for crest is GFN2-XTB//GFN-FF, with an energetic window
# ("--ewin") of 10 kcal/mol.
# opt> - the structures will be optimized at the global theory level
# (GFN2-xTB via TBLITE, ALPB(DMF) solvation).
# The optimized output will be written to ala_ala_crest_confs_opt.xyz.
# The freq> operator will perform a frequency calculation for each conformer,
# calculating free energies at the temperature (T_C) of
# -10 °C (default would be 25 °C).
# Each frequency calculation job will create an ORCA-like ".out" file.
2. Complex transition state search routine¶
CALC=UMA SOLVENT=CH2Cl2 NEB(IMAGES=9)
saddle> neb> scan> CH3Br_Br-.xyz 0 2
# The presence of one "-" in the structure filename will set its charge to -1.
# This can be always overridden by specifying an attribute in the molecule line:
# ...> ...> mol+++.xyz charge=0
# SOLVENT: The UMA model does not support implicit solvation natively.
# A ΔG_solv term will be added at the ALPB level via TBLITE.
# CH3Br_Br-.xyz was provided with two "reactive indices": 0 (Br) and 2 (C).
# scan> - A relaxed linear scan will be conducted between the two reactive indices.
# The input structure features no bond between index 0 (Br) and 2 (C), therefore
# the scan will try to form a bond between these two by reducing their distance.
# The scan will terminate when the atoms are appropriately close.
# This operator will return a file with all the scanned structures, passed to neb>.
# neb> - A NEB calculation will be set up with the scan structures, extracting the
# most spaced apart 9 images from the multi-xyz scan file. The default NEB mode will
# run a NEB-CI procedure, returning a transition state guess.
# saddle> - Will perform a saddle point optimization on the NEB transition state
# guess, and run a vibrational analysis on the converged structure.
3. Constrained conformational search, partial optimization, saddle optimization¶
CALC=UMA T=300 NCI
saddle> opt> crest_search> old_ts.xyz
B 34 144 # target: current distance
B 34 143 auto # target: current distance
B 1 2 2.345 # target: 2.345 Å
D 1 2 3 4 30.0 # target: 30°
# crest_search> A constrained conformational will be performed via CREST
# at the default GFN2-XTB//GFN-FF level (4 constraints), in NCI mode
# (NCI, analogous to the "--nci" keyword for CREST).
# opt> The resulting ensemble will be relaxed again with UMA,
# enforcing all 4 constraints.
# saddle> The structures will be optimized to a first order saddle point
# with no active constraints.
# Thermochemistry will be calculated at 300 K.
4. Atropisomer rotation¶
KCAL=5
scan> atropisomer.xyz 1 2 9 10
# scan> : (four indices specified) performs two dihedral
# scans (clockwise/anticlockwise) rotating the specified
# dihedral angle in 10° increments. Then, peaks above
# 5 kcal/mol (KCAL keyword, default 10) form the lowest energy
# structure are re-scanned at increased accuracy (1°
# increments). Maxima are returned and written to file.
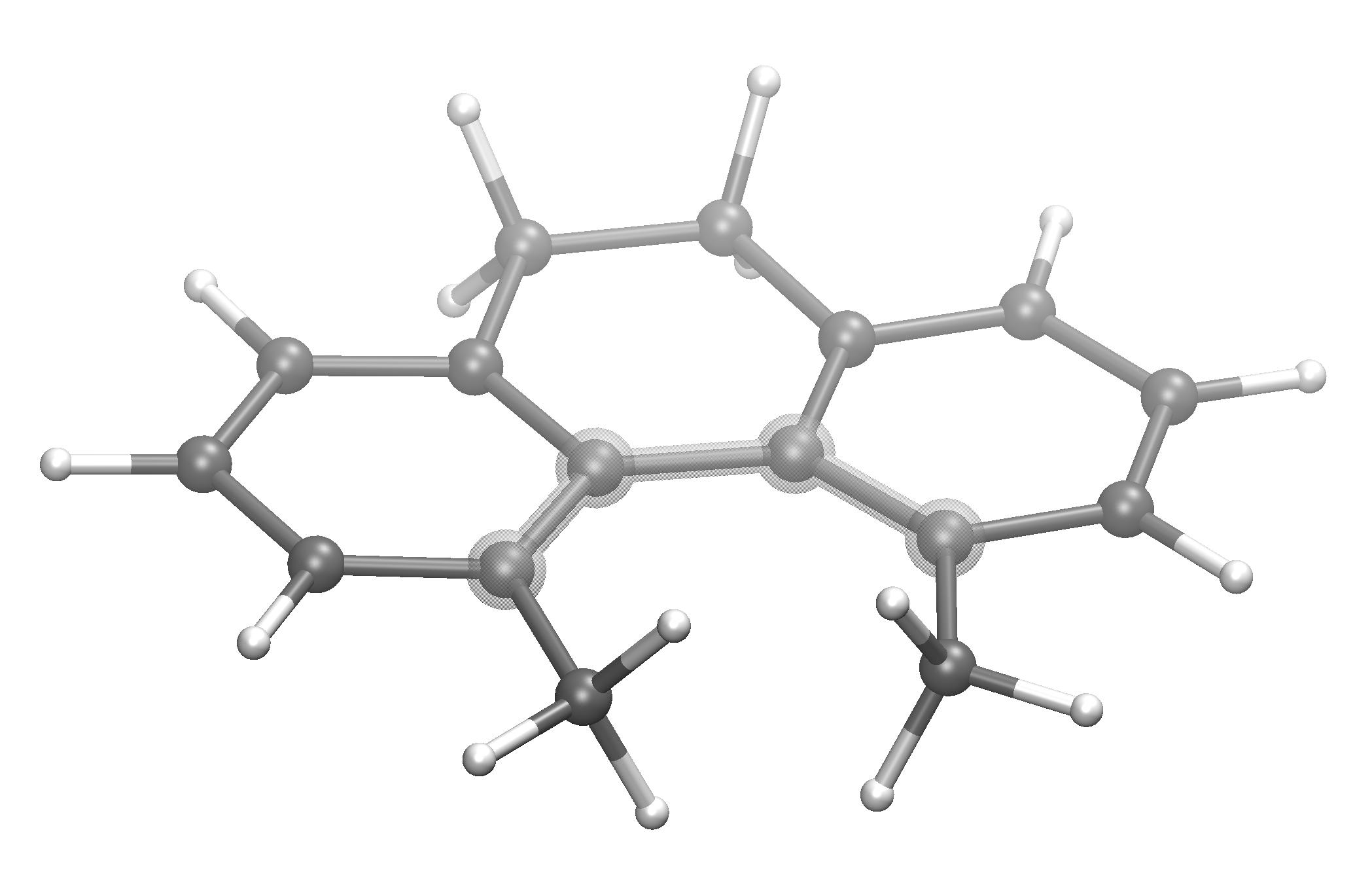
Best transition state arrangement found for the above input¶

Plot of energy as a function of the dihedral angle (part of the program output).¶