Legacy: Embedding runs¶
If the provided input is consistent with an embedding, one will be carried out. Embeddings (construction of molecular complexes) can be of six kinds:
dihedral - One molecule, four reactive atoms (i.e. racemization of BINOL)
string - Two molecules, one reactive atom each (i.e. SN2 reactions)
chelotropic - Two molecules, one with a single reactive atom and the other with two reactive atoms (i.e. epoxidations)
cyclical (bimolecular) - Two molecules, two reactive atoms each (i.e. Diels-Alder reactions)
cyclical (trimolecular) - Three molecules, two reactive atoms each (i.e. reactions where two partners are bridged by a carboxylic acid like the example above)
Reactive atoms supported include various hybridations of
C, H, O, N, P, S, F, Cl, Br and I. Many common metals are also
included (Li, Na, Mg, K, Ca, Ti, Rb, Sr, Cs, Ba, Zn), and it is easy
to add more if you need them (from reactive_atoms_classes.py).

Colored dots represent the imposed atom pairings.¶
Pairings¶
After each reactive index, it is possible to specify a pairing letter
representing the “flag” of that atom. If provided, the
program will only yield poses that respect these atom
pairings. It is also possible to specify more than one flag per atom,
useful for chelotropic embeds - i.e. the hydroxyl oxygen atom of a peracid, as
4ab.
Embeddings: good practice and suggested options¶
Here are some guidelines for conformational embedding. Not all of them apply to all embed types, but they will help in getting the most out of the program.
1) If a given molecule is present in the transition state, but it is not strictly involved in bonds breaking/forming, then that molecule can be pre-complexed to the moiety with which it is interacting. That is, the bimolecular complex can be used as a starting point. This can be the case for multimolecular adducts of non-covalently-bound catalysts.
2) Use the rdkit_search> or crest_search> operators, or provide conformational
ensembles obtained with other software.
3) FIRECODE default parameters are tentatively optimized to yield good results
for the most common situations. However, if you
have more information about your system, specifying details of the pairings
and options for your system is likely to give better results. For example,
embedding trimolecular TSs without imposed pairings generates about 8
times more structures than an embed with defined pairings. Also, if
reactive atoms distances in the transition state are known, using the
DIST keyword can yield structures that are quite close to
ones obtained at higher levels of theory. If no pairing
distances are provided, a distance guess is performed based on the atom type
(defaults are editable in the parameters.py file).
4) If FIRECODE does not find any suitable candidate for the given reacion,
you could try the SHRINK keyword (see keywords section).
Legacy: example embeddings¶
1. Trimolecular input¶
DIST(A=2.135)
maleimide.xyz 0A 5x
opt> HCOOH.xyz 4x 1y
crest_search> dienamine.xyz 6A 23y
# First pairing (A) is the C-C reactive distance
# Second and third pairings (x, y) are the
# hydrogen bonds bridging the two partners.
# Fixed constraints (A, UPPERCASE letters) will refine to the imposed values (here a=2.135 A)
# Interaction constraints (x, y, lowercase letters) will relax to an optimal value
# opt> - structure of HCOOH.xyz will be optimized before running the embedding
# crest_search> - A conformational search will be performed on dienamine.xyz before running the embedding
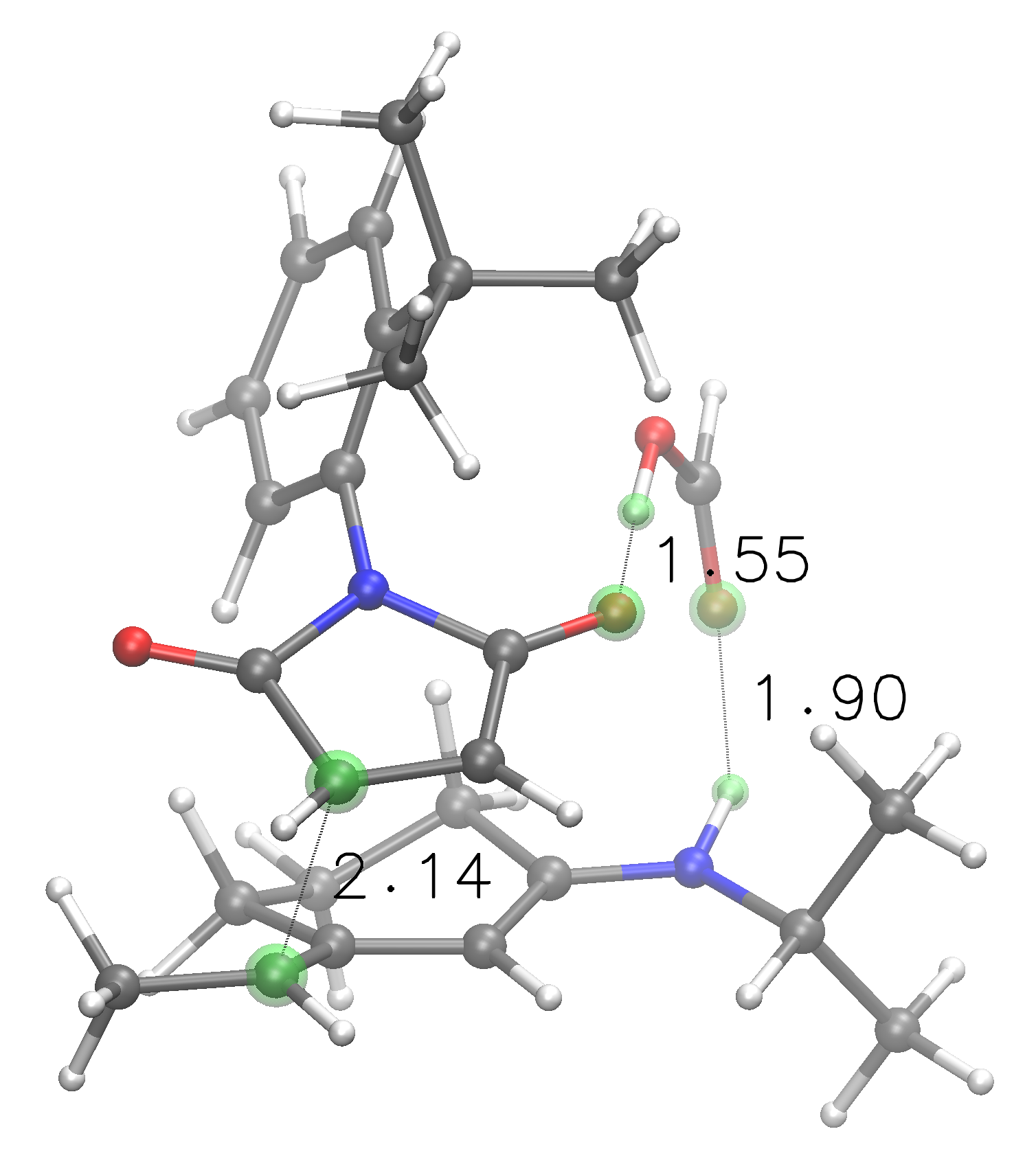
Best transition state arrangement found by FIRECODE for the above trimolecular input, following imposed atom spacings and pairings¶
2. Peptide-substrate binding mode¶
RMSD=0.3
crest_search> hemiacetal.xyz 34x
crest_search> peptide.xyz 39x
# Complex binding mode between a reaction
# intermediate (hemiacetal) and the catalyst
# (peptide).
# RMSD=0.3 reduces the similarity threshold to
# retain more structures (default 0.5 or 1 A)
# crest_search> performs a conformational
# search on hemiacetal.xyz (2 diastereomers,
# total of 72 conformers)
# String algorithm: 5.18 M poses checked
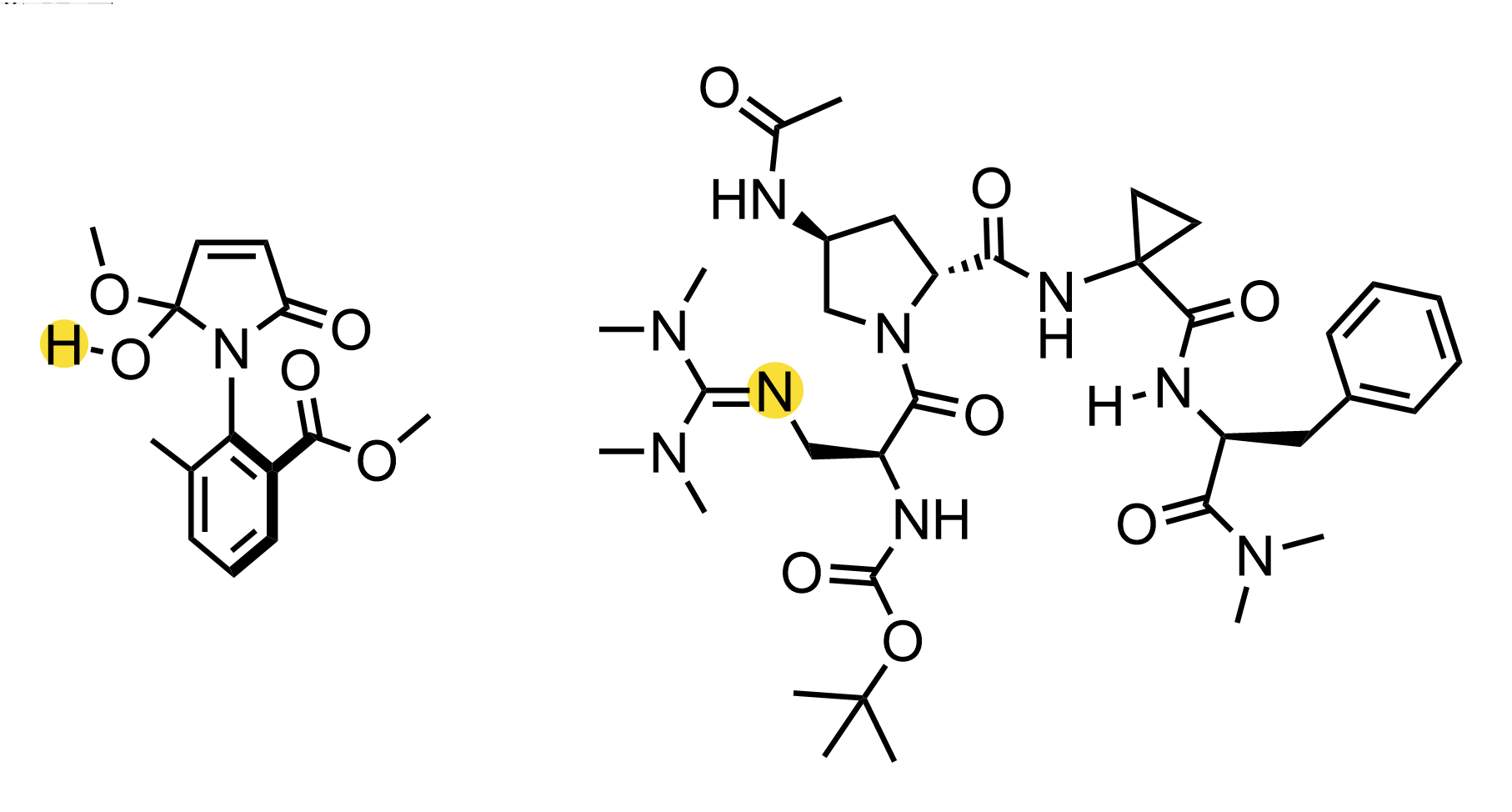
Input structures for hemiacetal.xyz (left) and peptide.xyz (right)¶
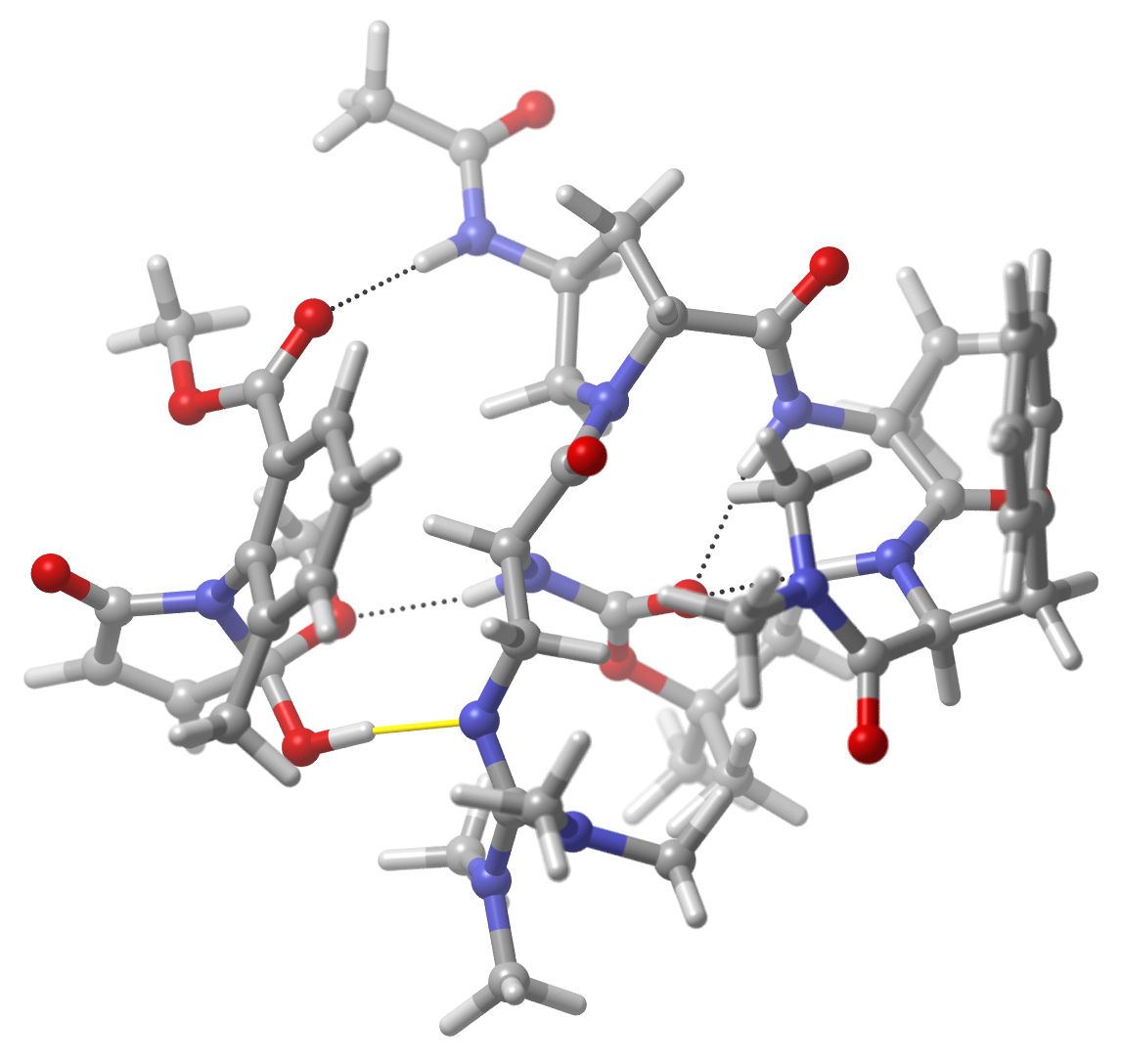
Best pose generated for the above input. The yellow bond is the imposed interaction, dotted lines are hydrogen bonds¶
3. Complex embedding with internal and external constraints¶
DIST(a=2.0, x=1.6, y=1.6) SOLVENT=ch2cl2 KCAL=20
crest_search> quinazolinedione.xyz 6A 14A 0x 7y
rdkit_search> peptide.xyz 0x 88y 19z 80z
# Four pairings provided (A, x, y, z):
# A - Fixed (UPPERCASE letter), internal to quinazolinedione
# (green) - kept at 2.0 Å during the entire run
# x - Interaction (lowercase letter) - will be embedded at 1.6 Å
# and then relaxed during the ensemble optimization steps (red)
# y - Interaction (lowercase letter) - will be embedded at 1.6 Å
# and then relaxed during the ensemble optimization steps (orange)
# z - Interaction (lowercase letter), internal to peptide (light blue)
# No distance provided, will relax during optimization
# crest_search> - metadynamics-based conformational search through CREST.
# Note that this is internal constraints-aware, and will treat the "A",
# "x", "y" and "z" pairings as bonds, retaining the specified distances.
# The KCAL keyword sets the energy threshold in kcal/mol for both the final
# ensemble and the metadynamics-based conformational search ("--ewin" in CREST).
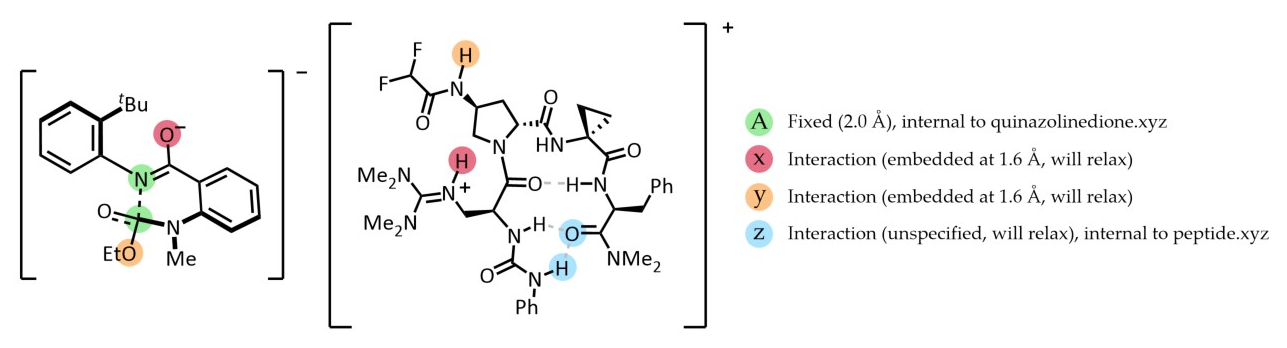
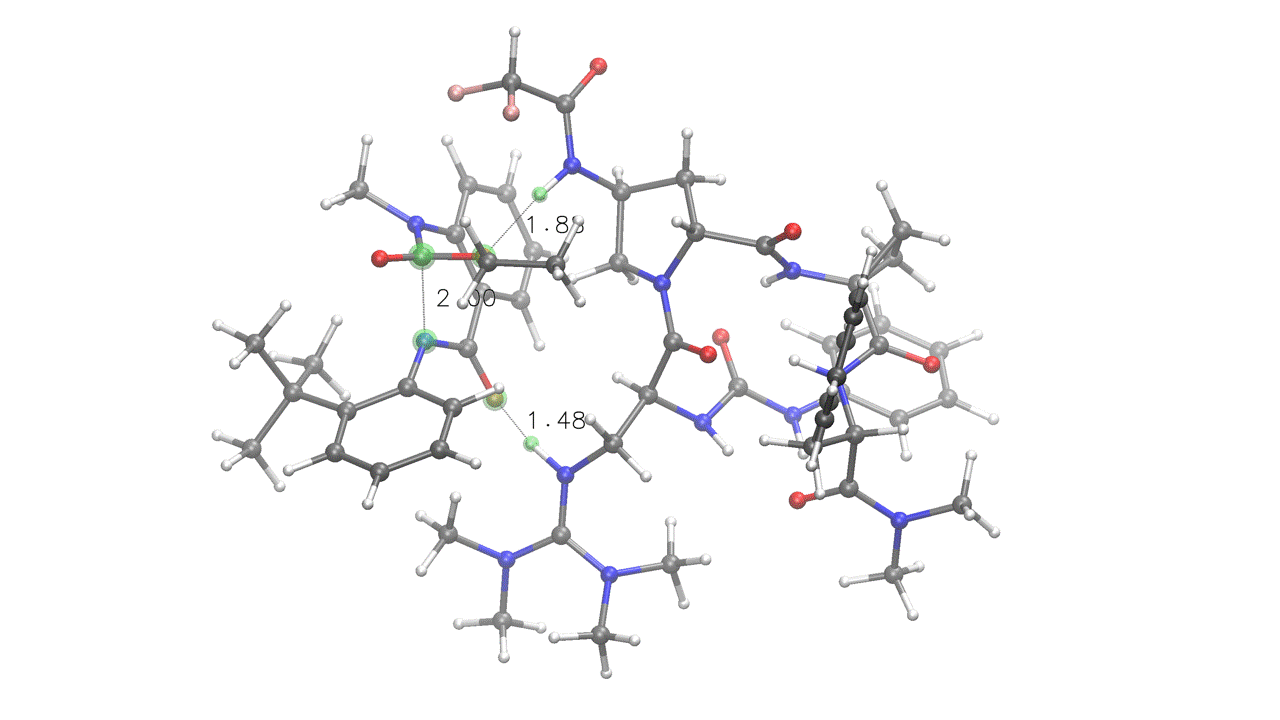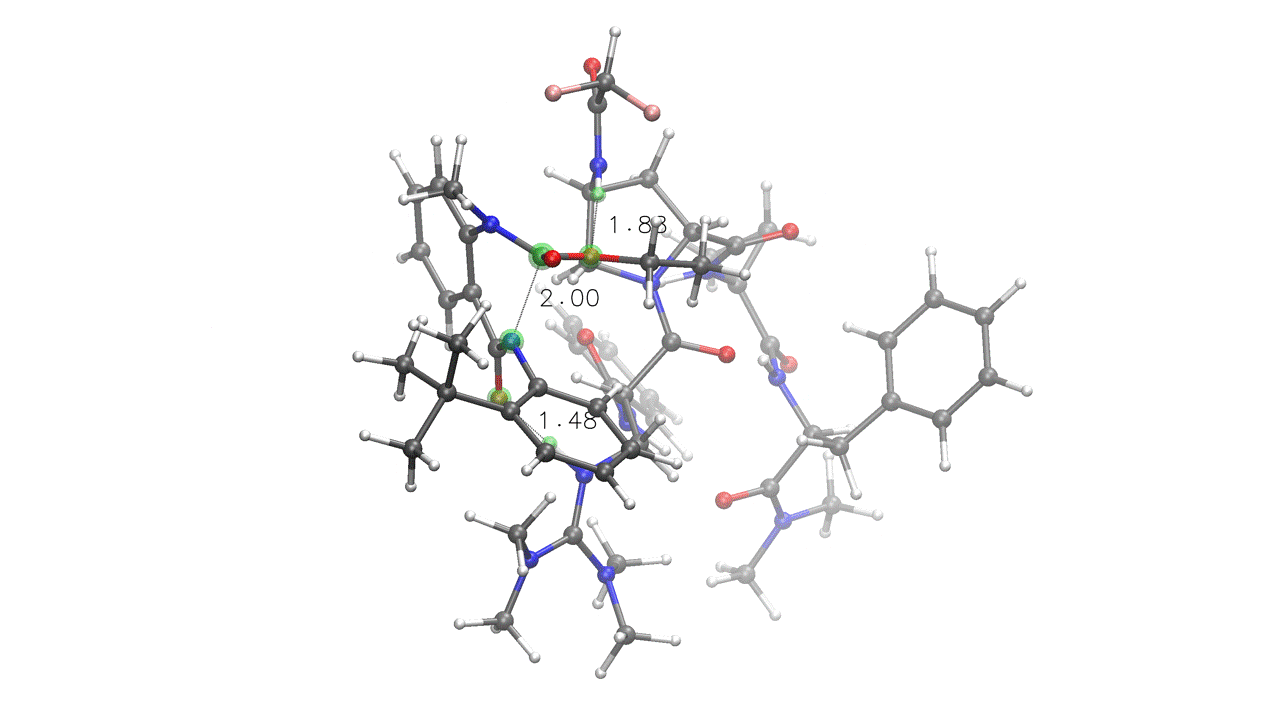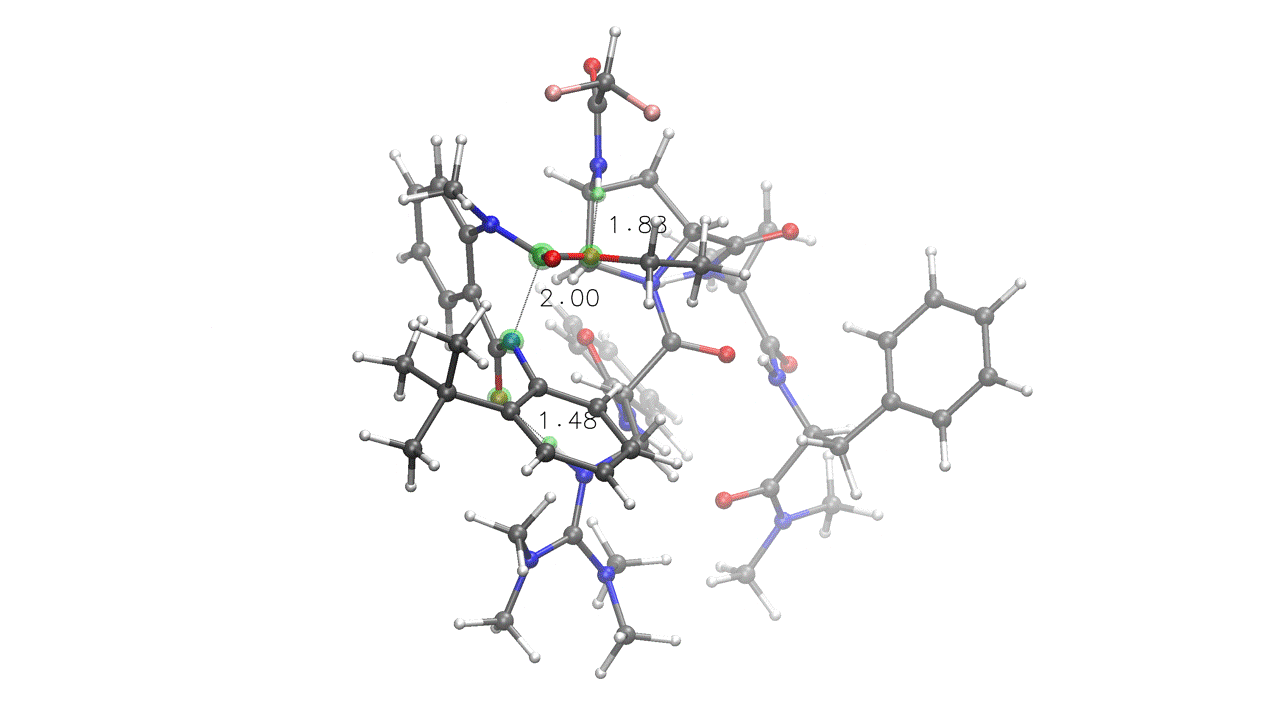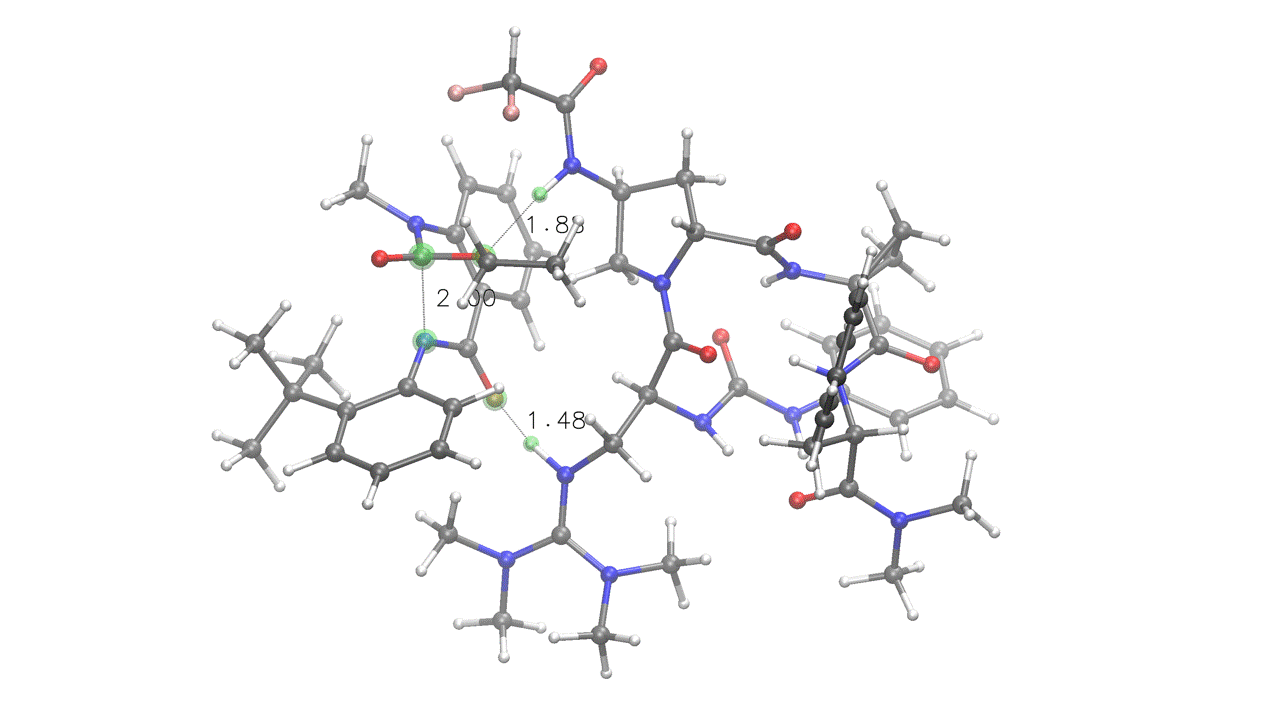
One of the poses generated for the above input. Note how fixed constraints were mantained (a=2) while interactions were relaxed (x=1.6, y=1.6, z)¶